Hierarchy Design for MMoCHi
Authors: Daniel Caron and William Specht
MMoCHi classification is centered around user-defined hierarchies. In this tutorial we will walk through how to set up a hierarchy and will give tips and recommendations for designing your own hierarchies.
Import packages
[1]:
import anndata
import scanpy as sc
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
plt.rcParams['figure.dpi'] = 150
plt.rcParams['figure.facecolor'] = 'white'
plt.rcParams['legend.frameon'] = 'False'
import mmochi as mmc
import os
mmc.log_to_file('hierarchy')
# Make the data directory if this notebook is not being run in the docs folder
if not os.path.isdir('data'):
os.mkdir('data')
[2]:
#global defaults
mmc.DATA_KEY = 'landmark_protein'
mmc.BATCH_KEY = 'batch'
Downloading and preprocessing the data
We will begin by downloading the data and performing preprocessing and landmark registration (see Integrated Classification tutorial):
[3]:
batches = ['pbmc_10k_protein_v3','5k_pbmc_protein_v3']
files = ['data/pbmc_10k_protein_v3.h5','data/5k_pbmc_protein_v3.h5']
urls = ['http://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_10k_protein_v3/pbmc_10k_protein_v3_filtered_feature_bc_matrix.h5',
'http://cf.10xgenomics.com/samples/cell-exp/3.0.2/5k_pbmc_protein_v3/5k_pbmc_protein_v3_filtered_feature_bc_matrix.h5']
adatas = mmc.utils.preprocess_adatas(files,
backup_urls=urls, log_CP_ADT=1e3, log_CP_GEX=1e4)
adata = anndata.concat(adatas, merge='first', keys=batches, label='batch', index_unique='_')
adata.obsm['protein'].drop(['IgG2a_control','IgG2b_control','IgG1_control'], axis=1, inplace=True)
adata = mmc.landmark_register_adts(adata, single_peaks=['CD25'])
Running with batch batch
Exploring the data
As described in the Integrated Classification tutorial, before cell type classification, we recommend performing a cursory unsupervised analysis of your dataset. This will help inform which cell types to include in your hierarchy and can be used to evaluate the quality of various cell type markers. As before, because clustering is stochastic, we will try to load in precomputed X_UMAP and leiden clusters:
[4]:
if os.path.isfile('data/Integrated_Classification_X_umap.txt') and os.path.isfile('data/Integrated_Classification_leiden.csv'):
adata.obsm['X_umap'] = np.loadtxt('data/Integrated_Classification_X_umap.txt')
adata.obs['leiden'] = pd.read_csv('data/Integrated_Classification_leiden.csv', index_col=0).astype(str).astype('category')
else:
print('No pre made files found, generating new leiden clusers')
sc.pp.highly_variable_genes(adata)
sc.pp.pca(adata)
sc.external.pp.harmony_integrate(adata, adjusted_basis='X_pca', key='batch',
verbose=False, max_iter_harmony=20)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
sc.tl.leiden(adata)
np.savetxt('data/Integrated_Classification_X_umap.txt', adata.obsm['X_umap'])
adata.obs['leiden'].to_csv('data/Integrated_Classification_leiden.csv')
protein_adata = anndata.AnnData(adata.obsm['landmark_protein'], adata.obs.copy())
protein_adata.obsm['X_umap'] = adata.obsm['X_umap'].copy()
[5]:
sc.pl.umap(adata,color=['batch','leiden'], s=25, sort_order=False, wspace =.5)
sc.pl.umap(adata,color=['CD3E','CD4','CD8A','IL7R','NKG7','CD19','JCHAIN','CD14'],
s=25, sort_order=False, cmap='Reds')
sc.pl.umap(protein_adata, color=['CD3','CD4','CD8a','CD127','CD56','CD19','CD14'],
s=25, sort_order=False, cmap='Blues')
... storing 'feature_types' as categorical
... storing 'genome' as categorical
... storing 'pattern' as categorical
... storing 'read' as categorical
... storing 'sequence' as categorical
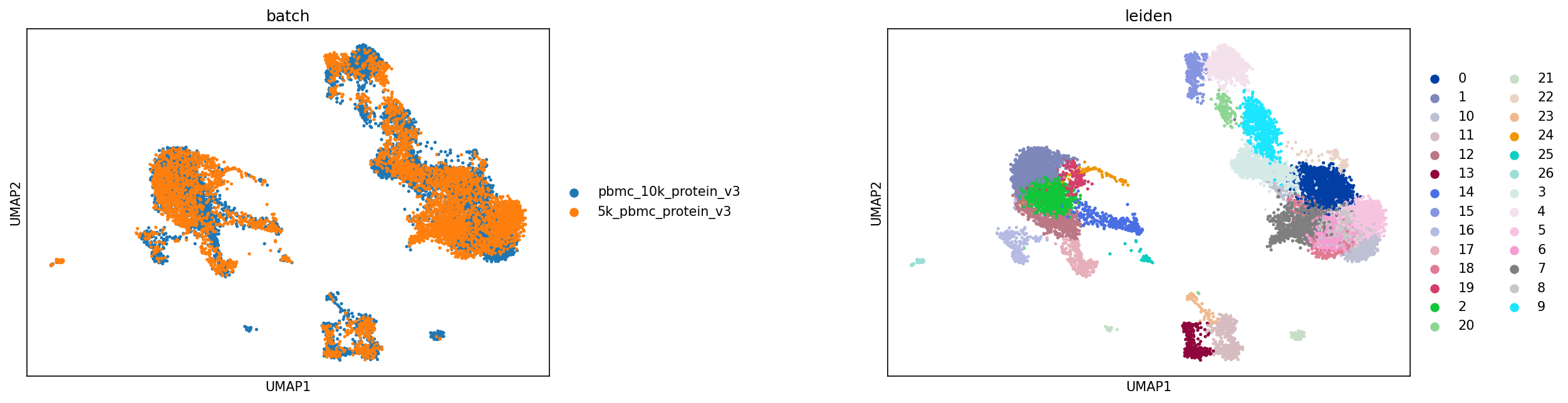
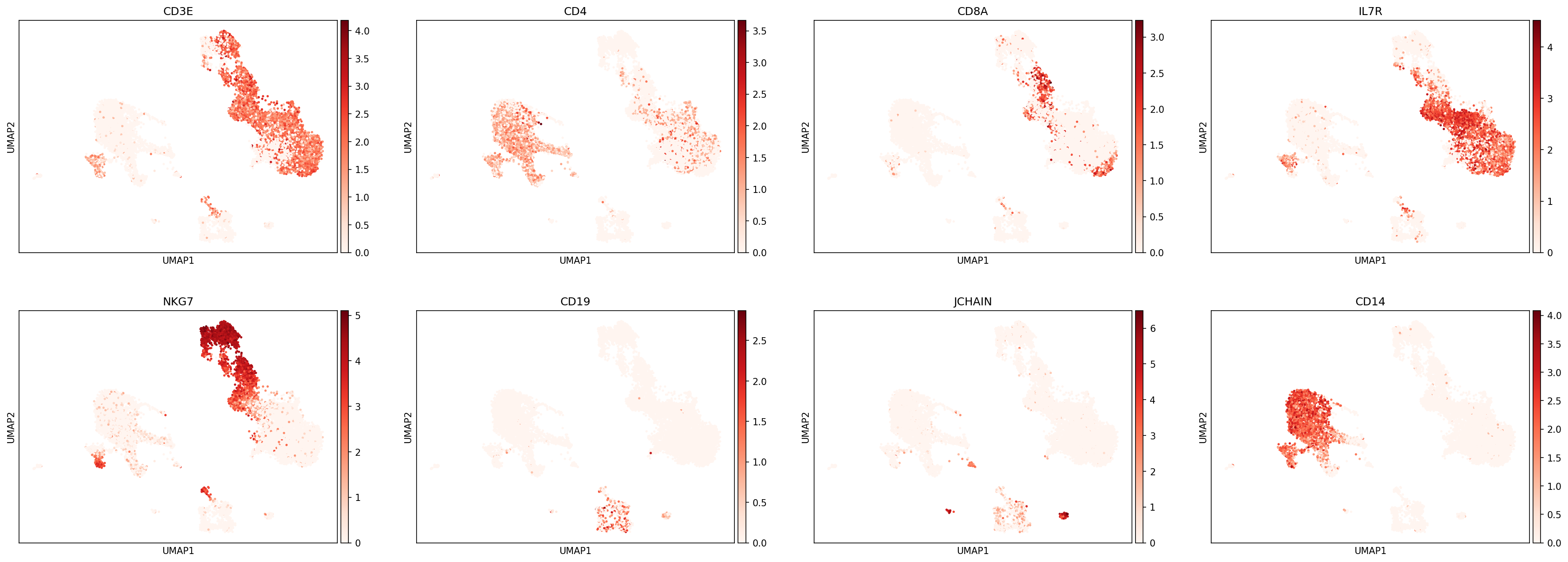
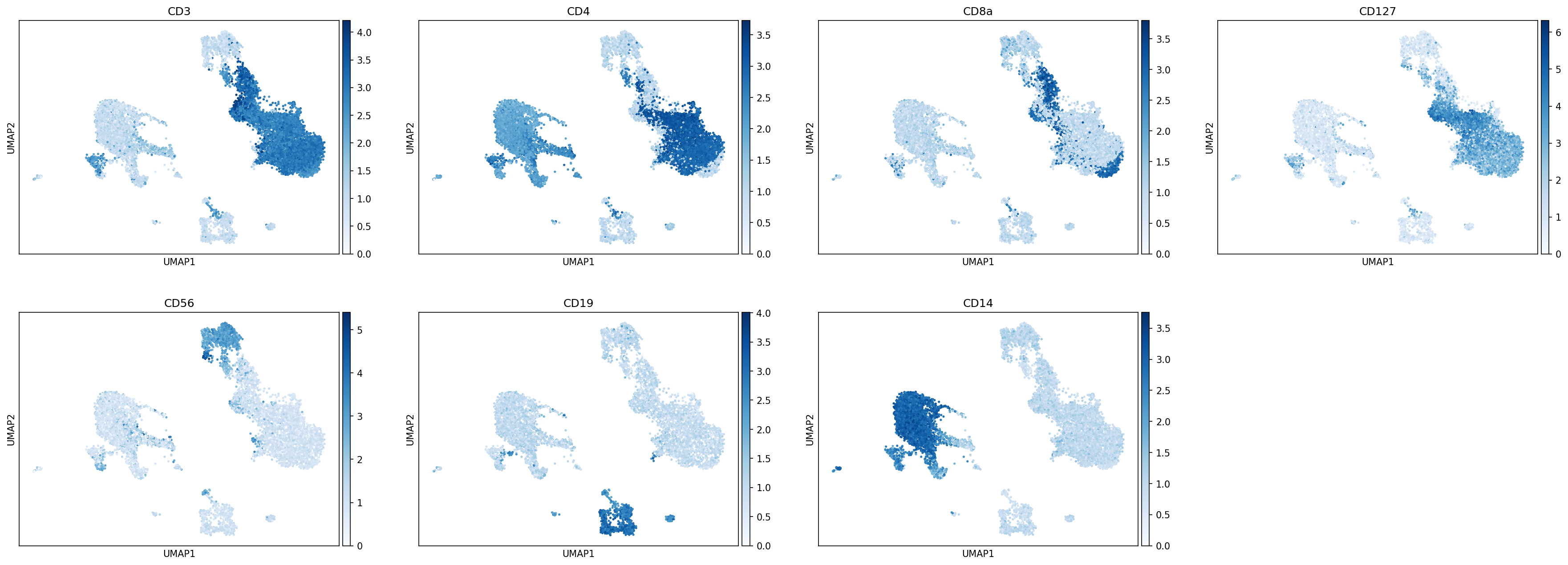
The first step in hierarchy design is defining your scope: What subsets would you like to annotate by classification?
MMoCHi classification and unsupervised clustering each have their own strengths and weaknesses. In our experience, integrated classification requires extra care to annotate subsets that are unique to individual batches, while unsupervised clustering can separate these events with ease. Moreover, we recommend a minimum of at least 100 events be captured by high-confidence thresholding for training with MMoCHi (although we have had some success with fewer training events). Thus, it may be
easier to focus on classifying cell types that are represented across multiple samples. For cell types that are better annotated by other methods, MMoCHi hierarchies support cutoff layers (using mmc.add_classification(is_cutoff=True)), which simply subsets cells by metadata or high-confidence thresholding without training a random forest.
Here, we could design a hierarchy encompassing various blood immune cell subsets, including Monocytes, CD4+ and CD8+ T cells, B cells, plasma cells, NK cells, and ILCs. Next, we look though our clusters to identify if any do not belong to one of these groups. Leiden cluster 23 appears to be T/B cell doublets specific to one batch. Although these could have been removed during QC, if you want to keep these subsets in the anndata.AnnData object for later analysis, we can label them in
the .obs for annotation by a cutoff layer.
[6]:
adata.obs['T_B doublets'] = adata.obs.leiden.isin(['23']).astype(str)
Choosing markers for your subsets
There are many resources detailing markers for cell types across many sample types. A few to start with include the Human Protein Atlas, PanglaoDB, CellMarker, and CellTypist encyclopedias. These resources primarily focus on transcriptomic markers, but manufacturers have also compiled resources for surface protein markers. These databases can help you identify less familiar subsets and guide marker selection. There are also many guides for identifying cell type marker genes from within your dataset using differential expression, including here and here. These can help you tune marker selection to your data and classify previously undescribed cell types.
Once you have a sense for what markers could be used to identify your cell types of interest, evaluate their expression in the data. Your main focus should be whether the marker is as specific for your cell type as you expect, and whether it is as ubiquitous as you expect. Due to vast differences in experimental design and sequencing technologies, markers useful in one study may not be as applicable to your dataset.
We have created a differential expression helper function to assist with the identification of marker genes. For example, here we will compare a cluster enriched in cytotoxic NK cells (NKG7+ CD3-) to a cluster enriched in a subset of cytotoxic T cells (NKG7+ CD3+) to identify other markers (beyond CD3) that distinguish them. This plot uses cmap='jet' which gives better distinction for where to draw thresholds.
[7]:
diff_expressed = mmc.identify_group_markers(adata, '4', '20', n_ups=11, n_downs=12)
... storing 'T_B doublets' as categorical
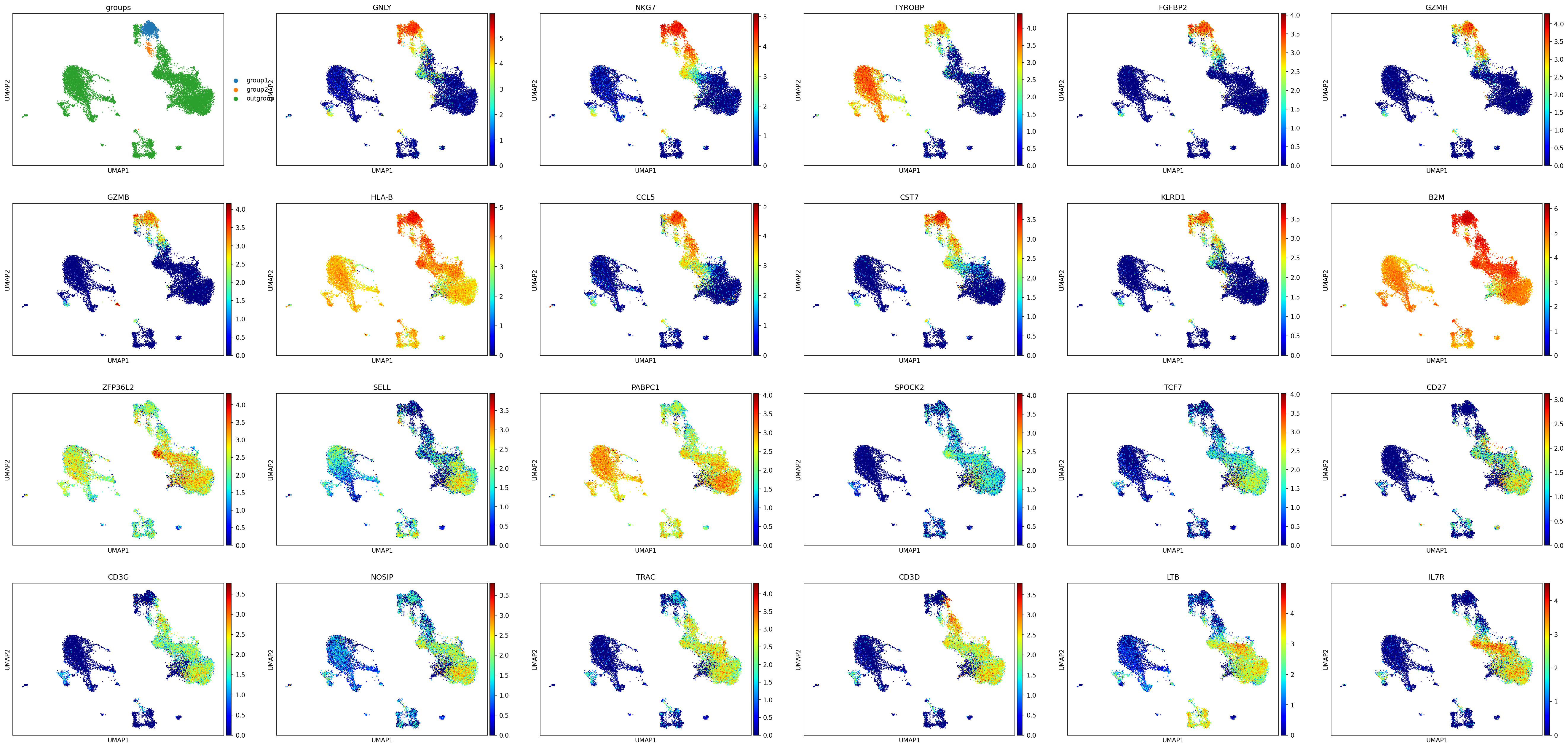
Here we can see that GNLY, TYROBP, and KRLD1 may be reliable transcriptomic markers of NK cells when properly thresholded. This also reveals that CD3G and CD3D may be more reliable T cell markers than CD3E, which is highly expressed within the NK clusters. We can also look at the results of the differential expression:
[8]:
diff_expressed[diff_expressed.names.isin(['GNLY','TYROBP','KLRD1','CD3G','CD3D'])]
[8]:
| names | scores | logfoldchanges | pvals | pvals_adj | |
|---|---|---|---|---|---|
| 0 | GNLY | 17.197853 | 1.553593 | 1.139587e-35 | 3.821946e-31 |
| 2 | TYROBP | 14.216040 | 2.010843 | 1.366718e-28 | 1.527899e-24 |
| 9 | KLRD1 | 11.747744 | 1.563995 | 1.054777e-22 | 2.721162e-19 |
| 33532 | CD3G | -9.948903 | -2.873073 | 8.484900e-18 | 1.293485e-14 |
| 33535 | CD3D | -11.975534 | -2.809434 | 9.826088e-24 | 3.295473e-20 |
Building the Hierarchy
We will begin by creating a new mmc.Hierarchy object. In the initialization of the object, we can set a lot of default parameters, which will affect how classification layers perform. For example, here we set default_min_events=15 so that MMoCHi will require at least 15 events to be used for training for each subset, and default_class_weight='balanced' to pass class_weight='balanced' to sklearn.ensemble.RandomForestClassifier(). Read more about various defaults that can
be set in the docs.
[9]:
h = mmc.Hierarchy(default_min_events=15, default_class_weight='balanced')
Defining a cutoff node
To build out the hierarchy, we will progressively define classification and subset nodes. To organize the hierarchy each node is defined with a unique name for itself and the name of its parent node.
First, we start with a classification node, which we will name 'Removal' and use for separating cells we wish to classify from those we don’t. This layer will be situated at the start of the hierarchy, directly beneath the root node (named All in the code). We also provide a list of features (generally gene or protein markers) for use during high-confidence thresholding. To access the cell metadata stored in the .obs during high-confidence thresholding, we simply provide the
column name (e.g. 'T_B doublets'). This layer is set to be a cutoff layer, which means that it will be used to directly subset cells, without training a random forest.
Next, we provide subset nodes, each with a name (e.g. 'Potential Doublets') and their parent (in this case, the 'Removal' node). Each subset node is also defined with a list corresponding to their expression of the markers provided, or in this case, their expected values in the .obs column. This can also be provided as a list of lists (to specify multiple satisfactory conditions) or as a dictionary of keyword arguments to pass to mmc.hc_defs for generation of a list of
lists.
[10]:
h.add_classification('Removal', 'All', ['T_B doublets'], is_cutoff=True)
h.add_subset('Potential Doublets','Removal', ['True'])
h.add_subset('To classify', 'Removal', ['False'])
Now let’s run this single-layer classifier and see the result!
[11]:
adata,h = mmc.classify(adata, h, 'lin', retrain = True, weight_integration=True)
adata = mmc.terminal_names(adata)
Setting up...
No provided modality name, defaulting to gex for all of .X
Using .X and landmark_protein
Removing 18757 features lacking expression in a minimum of 25 events...
Resorting to enforce sorted order of features by name
Set up complete.
Using 14795 features
Running with batch batch
Using weights of: [np.float64(0.4001677852348993), np.float64(0.5998322147651006)] for random forest n_estimators
Data subsetted on All in All
Running high-confidence populations for Removal...
Running high-confidence thresholds in 5k_pbmc_protein_v3
Running high-confidence thresholds in pbmc_10k_protein_v3
Performing cutoff for Removal...
Merging data into adata.obsm['lin']
Predicted:
Removal_class
To classify 13035
Potential Doublets 77
Name: count, dtype: int64
Converting columns in adata.obsm["lin"] to savable dtypes...
Great, looks like it worked correctly:
[12]:
pd.crosstab(adata.obs.classification, adata.obs['T_B doublets'])
[12]:
| T_B doublets | False | True |
|---|---|---|
| classification | ||
| Potential Doublets | 0 | 77 |
| To classify | 13035 | 0 |
A brief aside: defining a simple classification level
Lineage-level classification nodes tend to require many different markers to capture heterogenous populations. Before we dive into the next classification node, let’s start with a simpler example. Let’s say you have a dataset of only T cells, and are interested in selecting CD4 and CD8 T cells, based on expression of CD4 and CD8 protein, and CD4 and CD8A transcripts.
We will create an example hierarchy and add a classification level called 'Example level', specifying those markers. For convenience, marker names defined will be searched for using mmc.utils.get_data (see docs for details). For markers with unique names across the .var_names, .obsm[data_key].columns, and .obs.columns, we can just define them by that exact name, or use a unique pattern which occurs in that name. For markers with non-unique names, mmc.utils.get_data
will select the item from the .obsm[data_key].columns before searching the .var_names or .obs.columns. To bypass this automatic prioritization, users can append '_mod_data_key', '_gex', or '_obs' to the marker name. Additionally, to allow for the specification of multiple thresholds for a single marker (e.g. to select an intermediate expression), users can append '_hi' or '_lo' to the marker’s name. Here, you can see that we use both 'CD4' to select the
protein and 'CD4_gex' to specify the transcript.
Next, we will define subsets for CD4 and CD8 T cells, both with 'Example level' as their parent node. We can then specify expression levels using ‘pos’ (for positive), ‘neg’ (for negative) and ‘any’ (for no restriction on expression of this marker) in a list. Here we define CD4 T cells as [CD4+ CD8- CD4+ CD8A-] and CD8 T cells as [CD4- CD8+ CD4- CD8A+]. We can quickly visualize this using the .display() method.
[13]:
example_h = mmc.Hierarchy()
example_h.add_classification('Example level', 'All', ['CD4','CD8','CD4_gex','CD8A_gex'])
example_h.add_subset('CD4 T cell', 'Example level', ['pos','neg','pos','neg'])
example_h.add_subset('CD8 T cell', 'Example level', ['neg','pos','neg','pos'])
example_h.display()
All
└── *Example level*
├── CD4 T cell (CD4+ CD8- CD4_gex+ CD8A_gex-)
└── CD8 T cell (CD4- CD8+ CD4_gex- CD8A_gex+)
Specifying these subsets as positive for both the protein and transcript is quite restrictive, especially considering the low expression of CD4 transcript. We may be interested in specifying that the high-confidence cells express either the protein or transcript. We can do this by providing a list of ['pos','neg','any'] lists, each specifying a satisfactory condition for labeling events as high-confidence. For example, we could do the following:
[14]:
example_h = mmc.Hierarchy()
example_h.add_classification('Example level', 'All', ['CD4','CD8','CD4_gex','CD8A_gex'])
example_h.add_subset('CD4 T cell', 'Example level', [['any','neg','pos','neg'], ['pos','neg','any','neg']])
example_h.add_subset('CD8 T cell', 'Example level', [['neg','any','neg','pos'], ['neg','pos','neg','any']])
example_h.display()
All
└── *Example level*
├── CD4 T cell (CD8- CD8A_gex-) and [(CD4_gex+) or (CD4+)]
└── CD8 T cell (CD4- CD4_gex-) and [(CD8A_gex+) or (CD8+)]
Defining a random forest classification node
Great, now let’s return to our original example and repeat the process to define another classification node and subsets. This one will be named 'Broad Lineages', will run on 'To classify', and will use a mix of gene and protein markers for high-confidence thresholding. We then will supply subsets with names ('Lymphocyte' and 'Myelocyte'), both underneath 'Broad Lineages'. For these subsets, we would like to use fairly complex marker definitions (expression of any 1 or
any 2 of a list of markers), which would be hard to express as a list of lists (see comments). Thus, we have created a helper function to generate these complex rules (mmc.hc_defs), which is our preferred method for inputting definitions for subsets. To use it, pass a dictionary of keyword arguments specifying marker expression, using the keyword pos for any positive markers, neg for any negative markers and any_of for complex rules:
[15]:
h.add_classification('Broad Lineages', 'To classify', ['CD14','CD33','MARCO','MERTK','CD3','CD19','CD127','JCHAIN','LILRA4'])
# h.add_subset('Lymphocyte','Broad Lineages', [['neg', 'neg', 'neg', 'neg', 'pos', 'pos', 'any', 'any'],
# ['neg', 'neg', 'neg', 'neg', 'pos', 'any', 'pos', 'any'],
# ['neg', 'neg', 'neg', 'neg', 'pos', 'any', 'any', 'pos'],
# ['neg', 'neg', 'neg', 'neg', 'any', 'pos', 'pos', 'any'],
# ['neg', 'neg', 'neg', 'neg', 'any', 'pos', 'any', 'pos'],
# ['neg', 'neg', 'neg', 'neg', 'any', 'any', 'pos', 'pos']])
h.add_subset('Lymphocyte', 'Broad Lineages', dict(neg=['CD14','CD33','MARCO','MERTK'], any_of=['CD3','CD19','CD127','JCHAIN'], n=2))
In addition to selecting any 1 or any few markers, there is one other type of complex rule we may want to introduce, which is a list of multiple conditions to satisfy. This is supported by providing a list of marker sets to any_of, and a list of integers to n. There is an additional parameter for how to link these conditions together, with '&' (when all any_of conditions must be met) or '|' connectors (when only one of the any_of conditions must be met). Here we
want to specify Myelocytes must be negative for a suite of markers, and express (either CD14 or CD33) or (both MARCO and MERTK) or (LILRA4). Then, let’s observe the hierarchy to make sure our definitions are correct:
[16]:
h.add_subset('Myelocyte', 'Broad Lineages', dict(neg=['CD3','CD19','JCHAIN'], any_of=[['CD14','CD33'],['MARCO','MERTK'],['LILRA4']], n=[1,2,1], any_ofs_connector='|'))
h.display()
All
└── *Removal*
├── Potential Doublets (T_B doublets==True)
└── To classify (T_B doublets==False)
└── *Broad Lineages*
├── Lymphocyte (CD14- CD33- MARCO- MERTK-) and [2 of (CD3+ CD19+ CD127+ JCHAIN+)]
└── Myelocyte (CD3- CD19- JCHAIN-) and [1 of (CD14+ CD33+) or 2 of (MARCO+ MERTK+) or 1 of (LILRA4+)]
You can refer to the mmc.hc_defs docs for more details:
[17]:
help(mmc.hc_defs)
Help on function hc_defs in module mmochi.hierarchy:
hc_defs(marker_list: List[str], pos: Union[List[str], str] = [], neg: Union[List[str], str] = [], other: Union[List[str], str] = [], POS: str = 'pos', NEG: str = 'neg', OTHER: str = None, UNDEFINED: str = 'any', any_of: Union[List[str], List[List[str]]] = [], any_ofs_connector: str = '&', n: Union[List[int], int] = 1, ANY_OPTIONS: Union[List[List[str]], List[str]] = ['pos', 'any'])
Helper function for defining simple or complex gating strategies for high-confidence thresholding.
Parameters
----------
marker_list
All the markers in the group, usually set by the Classification node above. Markers can also have "_lo" or "_hi" appended
to them to specify multiple thresholds on the same marker in the same node.
pos
List of markers that must be positive
neg
List of markers that must be negative
other
List of markers that must be other
POS
Default value describing pos (should usually not be altered)
NEG
Default value describing neg (should usually not be altered)
OTHER
String or list of strings containing the value(s) to write for each marker in "other".
UNDEFINED
Default value describing any undefined markers (should usually not be altered)
any_of
Used to define more complex gating. Can be represented as a list of markers (a single grouping) or a list of these lists (multiple pairings), where some of the markers must meet a specified condition. By default one member of the list must be positive and list of lists are connected by logical 'or's
any_ofs_connector
In the case of any_of being a list of lists, whether to join them with an "&" for 'and' or a '|' for 'or' to create complex gating,
"&" for "at least 1 of [CCR7,SELL] AND at least 1 of [TCF7, MAL]"
'|' for "at least 1 of [CD19, CD20] OR at least 2 of [JCHAIN, CD138, CD34]"
n
When using "any_of", how many in the group must match the threshold (e.g. n=2 is "at least 2 of"), when any_of is a list of lists,
this can become a list of ints the same length
ANY_OPTIONS
Default values describing when the condition is statisfied and alternative (should ususally not be altered).
The alternative string is the filler when those markers are not part of the condition.
e.g. for "any two positive" you would use ['pos','any'] for "only two positive" you would use ['pos','neg']
Like other options, if any_of is a list of lists, this can also become a list of lists.
Tip — Subset definitions
For all classifications, it is generally useful to define any subset with at least one positive and one negative feature. Defining a subtype only negatively can be especially problematic, as both gene and (to a lesser degree) protein expression suffer from dropout, meaning expression of these markers may not be captured on all events.
Thresholding
We recommend building your hierarchy out one level at a time, thresholding and testing each level before building another. This is especially important when selecting thresholds for rare populations, whose expression of a marker might be hard to see on histograms of the entire dataset. Here we will subset to events intended for classification (as defined above). For more details on selecting thresholds, see the High-Confidence Thresholding tutorial.
[18]:
h.reset_thresholds()
if os.path.isfile('data/integrated_thresholds.csv'):
h.load_thresholds('data/integrated_thresholds.csv')
else:
print('Unable to load thresholds, generate thresholds yourself using h.run_all_thresholds')
Loaded thresholds.
[19]:
h.run_all_thresholds(adata[adata.obs.classification=='To classify'], mode='fancy rerun all')
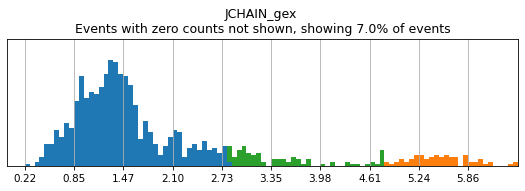
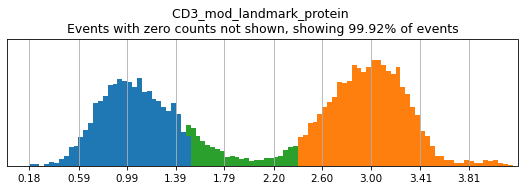
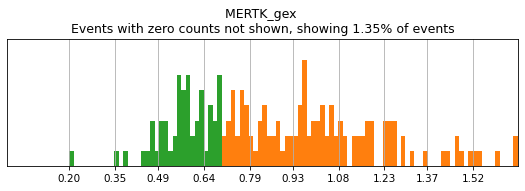
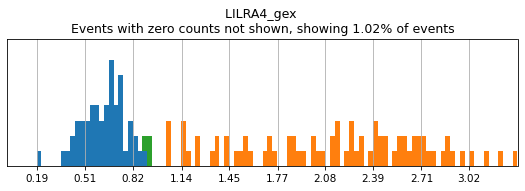
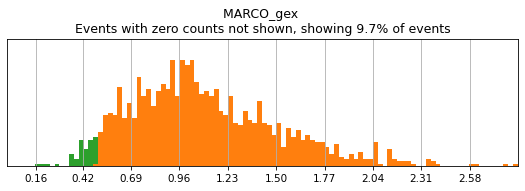
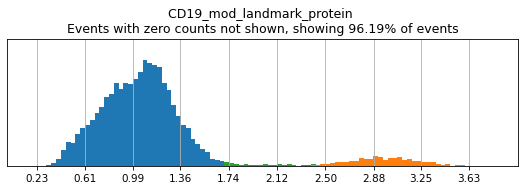
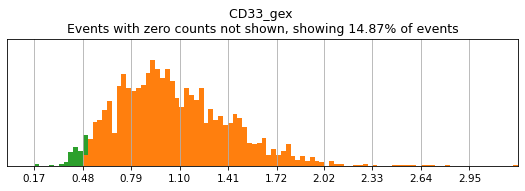
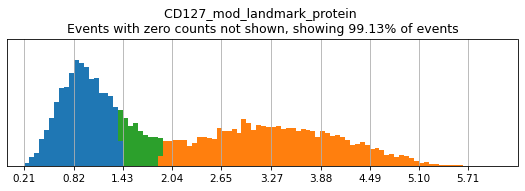
T_B doublets not found in adata, skipping thresholding.
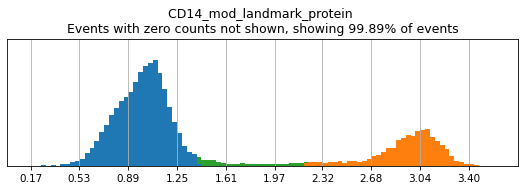
Completed!
Running classification
Now, let’s run classification at this level and evaluate the results! To prevent repetitively computing the classification, we have skip_to and end_at parameters that can be used to select which levels of the hierarchy should be rerun. This relies on information from previous runs being stored in the .obsm['lin']
[20]:
adata,h = mmc.classify(adata, h, 'lin', retrain=True, weight_integration=True, skip_to='Broad Lineages')
adata = mmc.terminal_names(adata)
Setting up...
No provided modality name, defaulting to gex for all of .X
Using .X and landmark_protein
Removing 18757 features lacking expression in a minimum of 25 events...
Resorting to enforce sorted order of features by name
Set up complete.
Using 14795 features
Skipped to classification on Broad Lineages
Running with batch batch
Using weights of: [np.float64(0.4001677852348993), np.float64(0.5998322147651006)] for random forest n_estimators
Data subsetted on To classify in Removal
Running high-confidence populations for Broad Lineages...
Running high-confidence thresholds in 5k_pbmc_protein_v3
Running high-confidence thresholds in pbmc_10k_protein_v3
Preparing training data for Broad Lineages...
Checking subsets for minimum events...
{'max_depth': 20, 'n_estimators': 100, 'n_jobs': -1, 'bootstrap': True, 'verbose': True, 'max_features': 'sqrt'}
Manually made a balanced class_weight: {'Lymphocyte': np.float64(1.2224023543512859), 'Myelocyte': np.float64(1.7387339866375937)}
Initializing classifier for Broad Lineages...
Training 41 new estimators using 5k_pbmc_protein_v3...
Choosing training data...
3020 real cells in training set...
Resampling...
/home/ubuntu/_Codebases/MMoCHi/mmochi/classifier.py:1077: FutureWarning: In the future, the default backend for leiden will be igraph instead of leidenalg.
To achieve the future defaults please pass: flavor="igraph" and n_iterations=2. directed must also be False to work with igraph's implementation.
sc.tl.leiden(adata,resolution = .5,n_iterations=1)
Found: 45 noise and 2 in danger of 3020 events.
Training with 3916 events after oversample resampling...
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 20 concurrent workers.
[Parallel(n_jobs=-1)]: Done 41 out of 41 | elapsed: 0.2s finished
Training 60 new estimators using pbmc_10k_protein_v3...
Choosing training data...
3521 real cells in training set...
Resampling...
Found: 84 noise and 0 in danger of 3521 events.
Training with 4766 events after oversample resampling...
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 20 concurrent workers.
[Parallel(n_jobs=-1)]: Done 10 tasks | elapsed: 0.1s
Merging data into adata.obsm['lin']
Running calibration on random forest
[Parallel(n_jobs=-1)]: Done 60 out of 60 | elapsed: 0.3s finished
Calibrating with method isotonic
[Parallel(n_jobs=20)]: Using backend ThreadingBackend with 20 concurrent workers.
[Parallel(n_jobs=20)]: Done 10 tasks | elapsed: 0.0s
Predicting for Broad Lineages...
[Parallel(n_jobs=20)]: Done 101 out of 101 | elapsed: 0.1s finished
[Parallel(n_jobs=20)]: Using backend ThreadingBackend with 20 concurrent workers.
[Parallel(n_jobs=20)]: Done 10 tasks | elapsed: 0.1s
Merging data into adata.obsm['lin']
Predicted:
Broad Lineages_class
Lymphocyte 9164
Myelocyte 3871
Name: count, dtype: int64
[Parallel(n_jobs=20)]: Done 101 out of 101 | elapsed: 0.6s finished
Converting columns in adata.obsm["lin"] to savable dtypes...
We can then use UMAPs or other metrics to evaluate the classifications at this level before proceeding:
[21]:
sc.pl.umap(adata, color=['batch','classification'], s=25, sort_order=False, wspace =.5)
sc.pl.umap(adata, color=['CD3E','CD4','CD8A','IL7R','NKG7','CD19','JCHAIN','CD14'],
s=25, sort_order=False, cmap='Reds')
sc.pl.umap(protein_adata, color=['CD3','CD4','CD8a','CD127','CD56','CD19','CD14'],
s=25, sort_order=False, cmap='Blues')
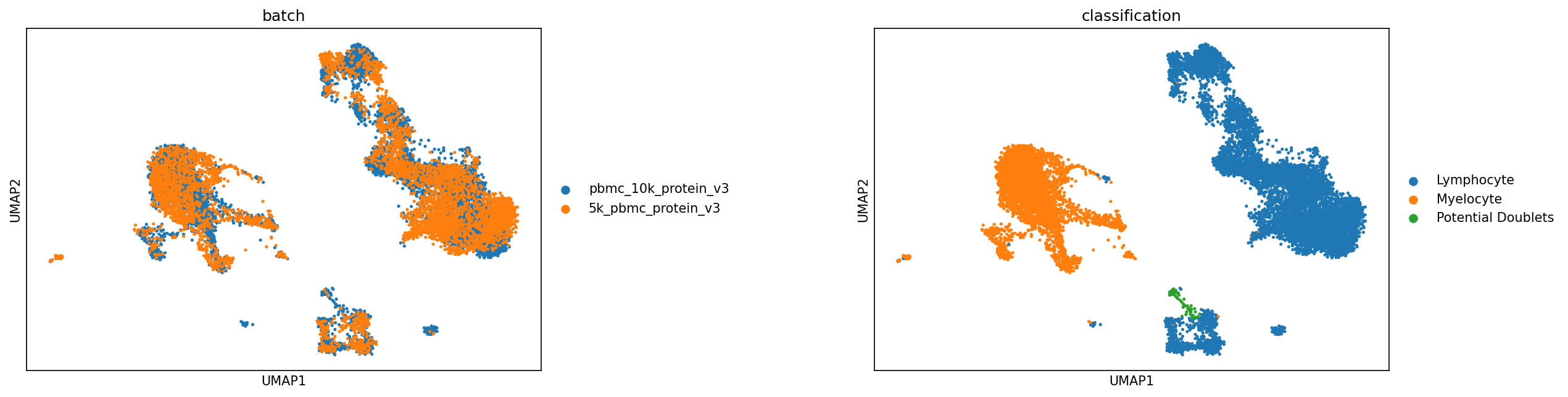
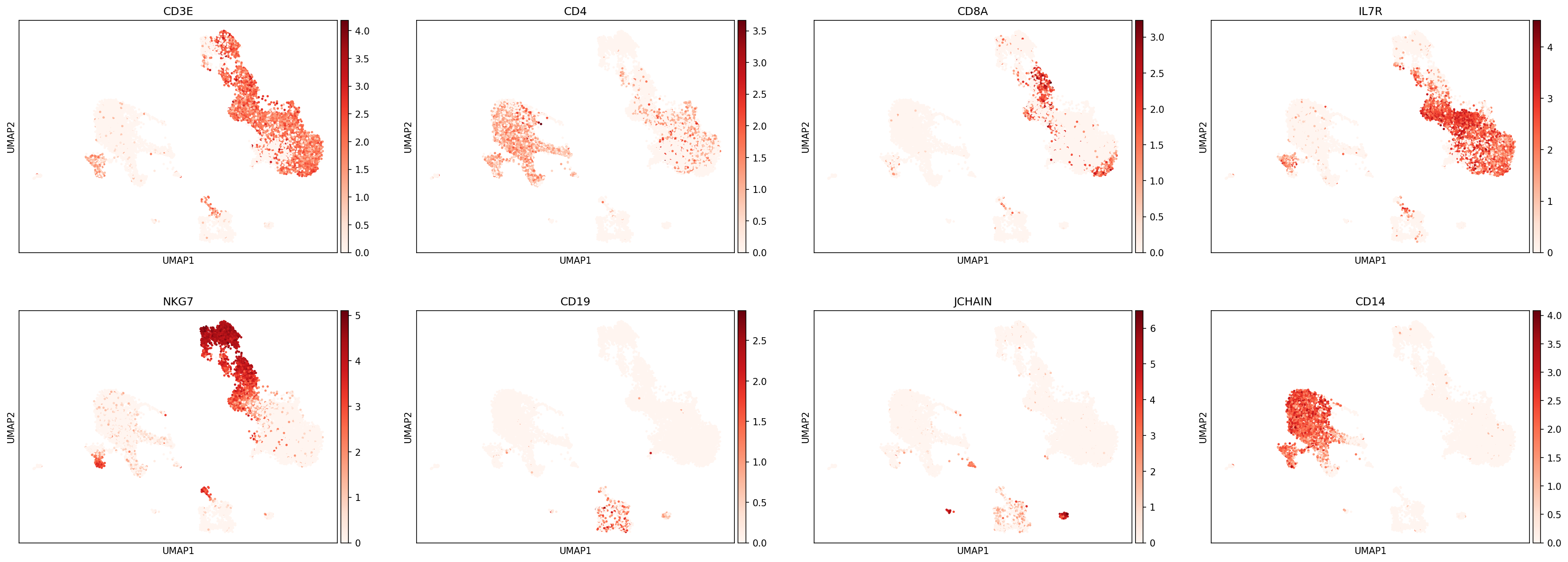

Defining more classification levels
Great, looks like that layer worked well. Now you can repeat this process for each additional classification layer until you have finished developing your hierarchy. We will discuss the remaining layers more broadly, rather than walking through each step of this process. We next defined classification levels for Lymphocyte subsets and CD4 and CD8 T cells. For lymphocyte subsets, we didn’t need to implement anything unique. For CD4 and CD8 T cells, we have tested classification and tuned
random forest hyperparameters (using clf_kwargs) to select more max_features at each level. Read more about kwarg options here.
[22]:
h.add_classification('Lymphoid', 'Lymphocyte', ['CD3','CD19','CD56','CD127','JCHAIN'])
h.add_subset('T cell', 'Lymphoid', dict(pos=['CD3','CD127'], neg=['CD19','JCHAIN']))
h.add_subset('B cell', 'Lymphoid', dict(any_of=['CD19','CD3','CD127','JCHAIN']))
h.add_subset('Plasma cell', 'Lymphoid', dict(any_of=['JCHAIN'], neg=['CD3','CD127']))
h.add_subset('NK_ILC', 'Lymphoid', dict(neg=['CD19','JCHAIN','CD3'], any_of=['CD127','CD56']))
h.add_classification('CD4_CD8', 'T cell', ['CD4','CD8','CD4_gex','CD8A_gex'], clf_kwargs=dict(max_features=.1))
h.add_subset('CD4 T cell', 'CD4_CD8', dict(neg=['CD8','CD8A_gex'], any_of=['CD4','CD4_gex']))
h.add_subset('CD8 T cell', 'CD4_CD8', dict(any_of=['CD8','CD8A_gex'], neg=['CD4','CD4_gex']))
Tip — Hierarchy design
Here, the hierarchy is setup to reflect known shared precursors and cell lineages, but lineage-based hierarchies aren’t always the most effective! When adding new cells to the hierarchy, consider what other cell types share the most transcriptomic and phenotypic identity. If two or more cell types share more features across an axis than they differ, it may be helpful to group them together and separate them lower in the hierarchy.
Once the hierarchy is created, you can use a helper function assign colors to each subset, and the hierarchy can be plotted using plot=True in the .display() method.
[23]:
h.color_dict(True, rot=1, hue=3, mode='DEPTH')
h.display(plot=True, font_mult=.75)

Tip — Subset colors
You can also define colors for subsets within each h.add_subset call. In that case, you can retrieve a dictionary of those colors for plotting purposes using h.color_dict().
Flattening a hierarchy
Sometimes, classification performance is low due to poor hierarchy design. This could be for many reasons, such as accidentally grouping two very distinct subsets, or performing subsetting in a nonoptimal order. If you would like to test whether your hierarchical design at any given level makes sense, you can try flattening individual levels using the h.flatten_children() method. This method allows children to inherent the definitions of their parents to make flattening a hierarchy a
bit easier. For example here, we could try classifying CD4 and CD8 T cells at the same time as other lymphocyte populations.
[24]:
h.flatten_children('T cell')
h.display(True, font_mult=.75)

Saving and loading a hierarchy
Once a hierarchy is created, it can be saved along with high-confidence thresholding and any random forest classification performed for application to a new dataset.
[25]:
h.save('data/ExampleHierarchy')
This hierarchy can then be loaded for training later
[26]:
h = mmc.Hierarchy(load='data/ExampleHierarchy')
Loading classifier from data/ExampleHierarchy...
Loaded data/ExampleHierarchy.hierarchy
Great!
Now you can create a hierarchy with complex expression dependencies!